الثلاسيميا
درست تانيا أونتربيرجر الصحافة وعلوم الاتصال في فيينا. في عام 2015 بدأت عملها كمحررة طبية في في النمسا. بالإضافة إلى كتابة النصوص المتخصصة ومقالات المجلات والأخبار ، يتمتع الصحفي أيضًا بخبرة في البث الصوتي وإنتاج الفيديو.
المزيد عن خبراء يتم فحص جميع محتويات بواسطة الصحفيين الطبيين.الثلاسيميا ، أو فقر الدم المتوسطي ، هو مرض وراثي يصيب خلايا الدم الحمراء. يتسبب الجين المعيب في أن ينتج الجسم القليل من صبغة الدم الحمراء (الهيموجلوبين) أو يتحلل بسرعة كبيرة. اعتمادًا على موقع الخلل الجيني ، يتم التمييز بين ثلاسيميا ألفا وبيتا. كلا الشكلين يؤدي إلى فقر الدم (فقر الدم). عادةً ما يعالجها الطبيب بالأدوية وعمليات نقل الدم. اقرأ المزيد عن الأسباب والأعراض والتشخيص والعلاج هنا!
رموز التصنيف الدولي للأمراض لهذا المرض: رموز التصنيف الدولي للأمراض هي رموز معترف بها دوليًا للتشخيصات الطبية. يمكن العثور عليها ، على سبيل المثال ، في خطابات الطبيب أو في شهادات العجز عن العمل. D57D56

لمحة موجزة
- الوصف: مرض محدد وراثيا لخلايا الدم الحمراء (كريات الدم الحمراء) يؤدي إلى فقر الدم.
- التشخيص: يقوم الطبيب بتشخيص مرض الثلاسيميا من خلال فحص دم خاص وتحليل المادة الوراثية (تحليل الحمض النووي).
- الأسباب: عيب جيني موروث يتسبب في إنتاج الجسم لصبغة دم حمراء قليلة جدًا أو معدومة (الهيموجلوبين).
- العلاج: العلاج هو الدواء ونقل الدم ، وفي بعض الحالات يكون زرع نخاع العظم ضروري.
- الأعراض: من بين أمور أخرى ، فقر الدم ، والتعب ، وتضخم الكبد والطحال ، واضطرابات النمو ، وتغيرات العظام ، وهشاشة العظام. غالبًا ما لا تظهر أي أعراض للأشكال الخفيفة من الثلاسيميا.
- الإنذار: الثلاسيميا لها درجات مختلفة من الشدة. كلما تم علاج الأعراض بشكل أسرع ، كان التشخيص أفضل. لا يمكن العلاج حاليًا إلا من خلال زرع الخلايا الجذعية أو العلاج الجيني.
ما هو مرض الثلاسيميا؟
التلاسيمية (أيضًا فقر الدم المتوسطي) هي مجموعة من الأمراض الوراثية التي يحدث فيها خلل في تكوين خلايا الدم الحمراء (كرات الدم الحمراء) بسبب خلل جيني. نتيجة لذلك ، ينتج الجسم القليل جدًا من صبغة الدم الحمراء (الهيموجلوبين) أو أنه لا ينتج عنها أبدًا أو يتحلل بسرعة كبيرة.
نتيجة لتغير الهيموجلوبين ، تكون خلايا الدم عادة أصغر من الطبيعي ولها عمر أقصر ، مما يؤدي إلى فقر الدم بمرور الوقت ، مصحوبًا بأعراض نموذجية مثل التعب وتسارع ضربات القلب.
يعتبر مرض الثلاسيميا من أكثر أمراض الدم شيوعًا لدى الأطفال والمراهقين. كما أنهم ينتمون إلى مجموعة اعتلالات الهيموغلوبين (اضطرابات الهيموغلوبين). يمكن أن ينتقل مرض الثلاسيميا من جيل إلى جيل. نظرًا لانتشار مرض الثلاسيميا بشكل خاص في منطقة البحر الأبيض المتوسط ، يُعرف أيضًا باسم "فقر الدم المتوسطي".
كيف يتطور مرض الثلاسيميا؟
الهيموجلوبين هو بروتين موجود في خلايا الدم الحمراء ويصنع في نخاع العظام. تمكن خلايا الدم الحمراء من حمل الأكسجين الحيوي من الرئتين إلى جميع مناطق الجسم.
يتكون الهيموغلوبين عادةً من أربع سلاسل بروتينية ، اثنتان منها متماثلتان - سلسلتان من نوع ألفا وسلاسل بيتا. يحتوي الهيموغلوبين أيضًا على الحديد الذي يربط الأكسجين. في حالة الثلاسيميا ، لا يشكل الجسم سلاسل بروتينية قليلة جدًا أو متغيرة (سلاسل ألفا أو بيتا) بسبب الجين المتغير (الطفرة).
هذا يعني أنه يمكن أن يتشكل عدد أقل من جزيئات الهيموجلوبين الوظيفية. تتقلص خلايا الدم الحمراء وتتحلل أكثر فأكثر. هذا يؤدي إلى فقر الدم ، حيث لم يعد الجسم مزودًا بالأكسجين بشكل كافٍ بسبب نقص خلايا الدم الحمراء.
اعتمادًا على سلسلة البروتين (سلسلة ألفا أو بيتا) التي تتأثر ، يتم التمييز بين ثلاسيميا ألفا (ألفا) وبيتا (بيتا) - ثلاسيميا.
ألفا ثلاسيميا
الشكل الأقل شيوعًا هو ثلاسيميا ألفا. في هذا الشكل ، يشكل الجسم عددًا قليلًا جدًا من سلاسل ألفا أو لا يتشكل على الإطلاق.
ثلاسيميا بيتا
ثلاسيميا بيتا هي الشكل الأكثر شيوعًا من الثلاسيميا. في هذا الشكل ، ينتج الجسم القليل جدًا من سلاسل هيموجلوبين بيتا أو لا ينتج عنها أبدًا.
يوجد أيضًا ثلاسيميا دلتا وجاما ، لكن كلاهما نادر جدًا وعادة ما يكون خفيفًا.
كيف يتم تشخيص مرض الثلاسيميا؟
نظرًا لأن أعراض الثلاسيميا غالبًا ما تظهر في مرحلة الطفولة ، فإن نقطة الاتصال الأولى عادة ما تكون مع طبيب الأطفال. إذا لزم الأمر ولإجراء مزيد من الفحوصات ، فسوف يحيلك إلى أخصائي في الطب الباطني (اختصاصي أمراض الدم).
تاريخ العائلة
نظرًا لأن الثلاسيميا مرض وراثي ، فعادة ما يوفر التاريخ العائلي للطبيب أدلة أولية. على سبيل المثال ، يسأل الطبيب ما إذا كان هناك حاملون معروفون للمرض في العائلة.
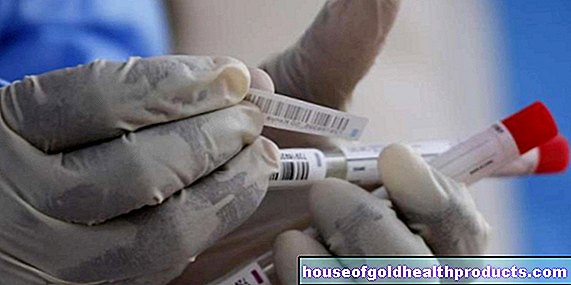
فحص الدم
لتأكيد التشخيص ، سيقوم الطبيب بإجراء فحص للدم بما في ذلك الرحلان الكهربي للهيموجلوبين (الهيموجلوبين الكهربائي).
الرحلان الكهربي للهيموجلوبين
مع الرحلان الكهربائي للهيموجلوبين ، يقوم الطبيب بإذابة الهيموجلوبين من دم المريض في سائل ويضعه على مادة حاملة خاصة (على سبيل المثال مصنوعة من الورق أو الجيلاتين). ثم يطبق الجهد.
اعتمادًا على كيفية تكوين الهيموجلوبين ، فإنه يتحرك مسافات مختلفة في وقت معين. بناءً على المسافة المقطوعة ، يقوم الطبيب بتقييم ما إذا كان الهيموجلوبين طبيعيًا أم معيبًا.
تحت المجهر ، يتعرف الطبيب عادة على الخلايا ذات الشكل المميز في دم الشخص. هذه هي خلايا الدم الحمراء الشاحبة نسبيًا مع ترسبات الهيموجلوبين ذات اللون الداكن في الوسط. خلايا الدم صغيرة ولديها هيموجلوبين ضعيف.
التشخيص عند الأطفال
يوصي الأطباء بإجراء فحص دم للأطفال حديثي الولادة الذين أصيبوا بالثلاسيميا (على سبيل المثال ، فحص حديثي الولادة). للقيام بذلك ، يأخذ الطبيب بعض الدم (على سبيل المثال من الحبل السري) بعد الولادة مباشرة ويفحصه بحثًا عن التغيرات الجينية. وبهذه الطريقة يمكن التعرف على المرض مبكرًا وعلاجه في الوقت المناسب.
التشخيص في الحمل
في الأطفال الذين لم يولدوا بعد ، يتم إجراء فحص الثلاسيميا (تشخيص ما قبل الولادة) باستخدام عينة نسيج من المشيمة.
يوصي الأطباء الآباء الحوامل الذين يعرفون أنهم حاملون للثلاسيميا بإجراء هذا التشخيص قبل الولادة في الأسابيع الاثني عشر الأولى من الحمل.
هل مرض الثلاسيميا وراثي؟
الثلاسيميا مرض وراثي. وهذا يعني أن المصابين بالثلاسيميا يرثون العيب الجيني من آبائهم ويولدون مصابين بالمرض.
الوراثة: ثلاسيميا ألفا
ألفا ثلاسيميا لها درجات مختلفة من الشدة. يتم التمييز بين alpha thalassemia minima و minmedia و intermedia و major. يعتمد التعبير على عدد الجينات المرضية التي ينقلها الوالدان إلى الطفل. هناك أربعة جينات لسلسلة ألفا ، اثنان من الأم واثنان من الأب.
يشارك ما مجموعه أربعة جينات في تكوين سلاسل ألفا. في ثلاسيميا ألفا الكبرى ، تكون جميع الجينات التي ورثها الطفل من الوالدين معيبة. وهو أشد أشكال الثلاسيميا ألفا. نادرًا ما يتمكن الأطفال المصابون بهذا الشكل من البقاء على قيد الحياة وعادة ما يموتون قبل الولادة أو بعد ذلك بأيام قليلة. فقط في حالات قليلة يمكن إبقاء الأطفال على قيد الحياة بمساعدة زرع الخلايا الجذعية.
الأشخاص المصابون بألفا ثلاسيميا الصغرى أو الوسيطة ورثوا اثنين أو ثلاثة من الجينات المعيبة من والديهم. وهي من بين الأشكال الأكثر اعتدالًا من الثلاسيميا ألفا وعادة ما تسبب أعراضًا معتدلة أو خفيفة أو لا تسبب أي أعراض لدى المصابين.
إذا ورث أحد الوالدين جينًا معيبًا لطفلهما ، فإن هذا يسمى ألفا ثلاسيميا الدنيا. وعادة لا يعاني المصابون من أعراض ويعيشون دون إعاقة.
الوراثة: ثلاسيميا بيتا
كما أن ثلاسيميا بيتا لها درجات متفاوتة من الشدة. وهي مقسمة إلى ثانوية ، وسيطة ، وكبرى.
الثلاسيميا الكبرى (وتسمى أيضًا فقر الدم في كولي) هي أشد أشكالها. يحدث عندما ينقل كلا الوالدين العيب الجيني إلى الطفل.
في حالة الثلاسيميا بيتا الكبرى ، من الضروري تبادل دم الشخص المصاب بانتظام (حوالي كل ثلاثة إلى أربعة أسابيع) عن طريق الحقن. إذا تُرك دون علاج ، فعادة ما يصاب المصابون بتشوهات شديدة في العظام. الأطفال المصابون هم أيضًا أكثر عرضة للإصابة بالعدوى ، فهم ضعفاء وعادة لا يتطورون وفقًا لأعمارهم.
بيتا ثلاسيميا الوسيطة هي شكل وسيط. ورث الأشخاص المصابون بهذا الشكل أيضًا الجينات المعيبة من كلا الوالدين. عادة ما تكون أعراضهم غير واضحة تمامًا كما في الشكل الرئيسي. ما عليك سوى نقل الدم في بعض الأحيان.
في ثلاسيميا بيتا الصغرى ، ورث المصابون جينًا واحدًا من سلسلة هيموغلوبين بيتا المعيب من أحد الوالدين. الوالد الآخر نقل الجين العامل إلى الطفل.
لا يعاني الأشخاص المصابون بالثلاسيميا بيتا الصغرى عادةً من أعراض فقر الدم الخفيفة. كقاعدة عامة ، لا يحتاجون إلى علاج. ومع ذلك ، وبصفتهم حاملين للجينات ، فإنهم قادرون على نقل مرض الثلاسيميا إلى أطفالهم.
إذا كان كلا الوالدين مصابين بالثلاسيميا بيتا الصغرى ، فهناك احتمال بنسبة 25٪ أن يرث الطفل الطفرات وأن يصاب بالثلاسيميا الكبرى أو المتوسطة. هناك احتمال بنسبة 50 في المائة أن يرث الطفل شكلاً ثانويًا وفرصة بنسبة 25 في المائة أن يكون الطفل بصحة جيدة.
من أجل الحصول على ثلاسيميا بيتا ، يجب على كلا الوالدين نقل جيناتهم المعيبة إلى الطفل. حامل المرض لديه جين معيب واحد فقط ، والجين الآخر وظيفي. على الرغم من أنهم لا يمرضون ، يمكنهم نقل الجين إلى أطفالهم.
أين يحدث مرض الثلاسيميا؟
يُعرف مرض الثلاسيميا أيضًا بالعامية باسم فقر الدم المتوسطي ، حيث ينتشر في دول البحر الأبيض المتوسط مثل إيطاليا واليونان. ولكنه يحدث أيضًا في الشرقين الأدنى والأوسط وفي أجزاء من إفريقيا وآسيا.انتشر مرض الثلاسيميا في أوروبا الوسطى من خلال روابط التجارة العالمية والهجرة.
منذ الستينيات ، ظهرت الثلاسيميا أيضًا في وسط وشمال أوروبا (مثل ألمانيا والنمسا وفرنسا وإنجلترا وهولندا وبلجيكا والدول الاسكندنافية). في غضون ذلك ، تعتبر الثلاسيميا من أكثر أمراض الدم شيوعًا لدى الأطفال والمراهقين.
ما مدى انتشار المرض؟
تشير التقديرات إلى أن حوالي 500 إلى 600 شخص في ألمانيا يعانون من شكل حاد من مرض الثلاسيميا. يعاني حوالي 200000 شخص في ألمانيا من النوع الخفيف من مرض الثلاسيميا. بالإضافة إلى ذلك ، تعتبر ثلاسيميا بيتا أكثر شيوعًا من ثلاسيميا ألفا.
كيف تعالج مرض الثلاسيميا؟
عادة ما يعالج الطبيب المتخصص (مثل أخصائي أمراض الدم لدى الأطفال) مرض الثلاسيميا اعتمادًا على شدة المرض والأعراض التي تحدث. يتم العلاج في المراكز المتخصصة في هذا المرض. اعتمادًا على ما إذا كان الشخص المصاب يحتاج إلى نقل دم ، يتم الآن التمييز بين العلاج المعتمد على نقل الدم (TDT) والعلاج غير المعتمد على نقل الدم (NTDT) لعلاج الثلاسيميا.
العلاج غير المعتمد على نقل الدم
عادة لا تتطلب الأشكال الخفيفة من الثلاسيميا علاجًا. ومع ذلك ، في حالات نادرة أثناء الحمل ، قد تصاب النساء الحوامل بفقر دم حاد بسبب الثلاسيميا الخفيفة. ثم يحتاجون بعد ذلك إلى عمليات نقل دم منتظمة لحماية الأم والطفل.
العلاج المعتمد على نقل الدم
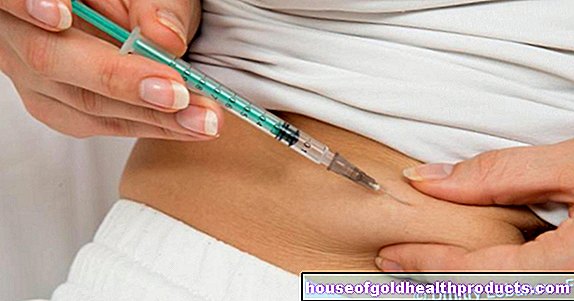
يقوم الطبيب بتبادل دم الأشخاص المعتمدين على نقل الدم (مثل الثلاسيميا الكبرى أو الوسطاء) مع عمليات نقل الدم المنتظمة. اعتمادًا على شدة مرض الثلاسيميا ، يحدث هذا عادةً كل ثانية إلى أسبوع ثالث ، وعادةً مدى الحياة.
غالبًا ما تؤدي عمليات نقل الدم الإضافية إلى زيادة الحديد. في هذه الحالات ، سيصف الطبيب دواء يزيل الحديد الزائد من الجسم (ما يسمى بالمخلبات أو خالب الحديد). يقوم الطبيب بحقنها تحت الجلد (تحت الجلد) في المستشفى أو يأخذ الشخص المعني أقراصًا في المنزل.
زرع الخلايا الجذعية
من الممكن علاج الثلاسيميا الكبرى بزرع الخلايا الجذعية. ومع ذلك ، يفترض هذا وجود متبرع مناسب للشخص المعني.
العلاج الجيني
يوجد علاج جيني لثلاسيميا بيتا المعتمد على نقل الدم منذ عام 2019. يعالج العلاج الخلل الجيني السببي ويمنح المصابين فرصة للعيش دون عمليات نقل الدم مدى الحياة.
في هذا العلاج الجديد ، يتم حقن جين سليم قادر على إنتاج الهيموجلوبين السليم في الخلايا الجذعية لدم الشخص. للقيام بذلك ، يتم أخذ الخلايا الجذعية أولاً من نخاع عظم المريض ومعالجتها بدواء. ثم يتلقى الشخص المعني الخلايا الجذعية الخاصة به (مع الجينات السليمة) مرة أخرى ، والتي تشكل الآن خلايا دم حمراء صحية وصبغة دم طبيعية.
وافقت وكالة الأدوية الأوروبية (EMA) على العلاج للأشخاص الذين تزيد أعمارهم عن 12 عامًا والذين يتناسبون مع هذا النوع من العلاج. العلاج الجيني متاح حاليًا فقط في ألمانيا. يتم علاج عدد قليل جدًا من المرضى حاليًا به.
النظام الغذائي ونمط الحياة
يمكن للأشخاص المصابين بالثلاسيميا التأثير بشكل إيجابي على مسار المرض من خلال تناول النظام الغذائي الصحيح واعتماد أسلوب حياة مناسب. يرجى ملاحظة ما يلي:
- تناول نظامًا غذائيًا متوازنًا.
- تجنب الأطعمة التي تحتوي على نسبة عالية من الحديد (مثل الكبد).
- احصل على ما يكفي من الكالسيوم من الطعام (مثل الحليب واللبن والجبن وأوراق السبانخ والبروكلي).
- لا تشرب الكحول.
- مارس التمارين الرياضية بانتظام (حوالي ثلاث إلى أربع ساعات من التمارين مثل المشي السريع أو الركض أو ركوب الدراجات في الأسبوع).
ما هي أعراض مرض الثلاسيميا؟
تعتمد شدة الأعراض على مقدار تغير الهيموجلوبين بشكل غير طبيعي. عادة ما يعاني الأشخاص المصابون بنوع خفيف من الثلاسيميا من أعراض قليلة أو معدومة.
ومع ذلك ، إذا لم يتم علاج الأطفال المصابين بنوع حاد من الثلاسيميا ، فإنهم يظهرون مشاكل صحية في وقت مبكر من عمر أربعة أو خمسة أشهر.
فقر دم
يُظهر الأشخاص المصابون بالثلاسيميا الكبرى أو الوسطية الأعراض النموذجية لفقر الدم في البداية:
- تكون شاحبة أو ذات جلد مصفر.
- أنت متعب طوال الوقت.
- تشعر بالدوار.
- لديك صداع.
- لديك ضربات قلب سريعة.
- سرعان ما يصابون بضيق التنفس أثناء النشاط البدني.
- غالبًا ما يكون لديهم تضخم في الكبد والطحال.
- غالبًا ما يتم إزعاج تطورهم.
للوهلة الأولى ، تشبه الثلاسيميا فقر الدم الناجم عن نقص الحديد. ومع ذلك ، فإن مستوى الحديد يكون منخفضًا في حالة نقص الحديد ، بينما يكون عادةً طبيعيًا في مرض الثلاسيميا.
في وقت لاحق أو إذا كان العلاج غير كافٍ ، غالبًا ما يعاني المصابون من آثار جانبية شديدة. وتشمل ، على سبيل المثال:
- تشوهات العظام
- عدوى متكررة
- مشاكل قلبية
- السكرى
- هشاشة العظام
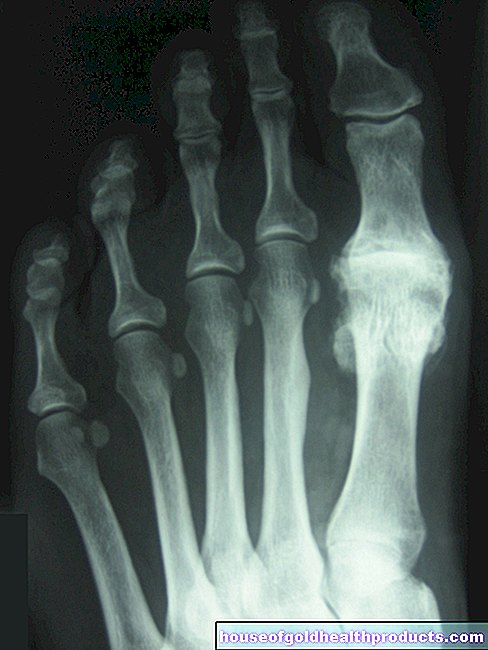
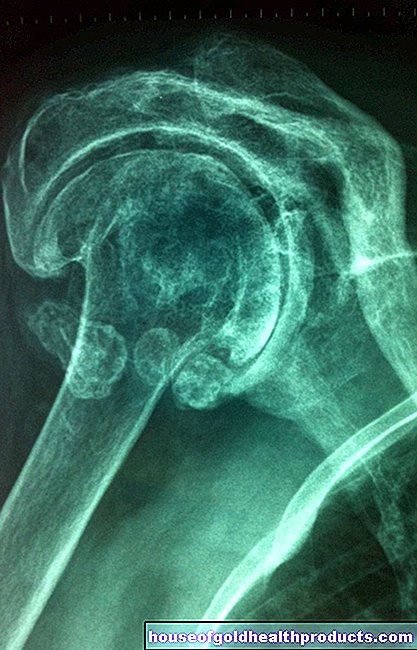
تغييرات العظام
غالبًا ما يُظهر الأشخاص المصابون بالثلاسيميا الكبرى أو المتوسطة نموًا غير طبيعي للعظام:
- تتضخم عظام الجمجمة.
- انتفاخ الجبهة والفك العلوي والعظم الوجني.
- ثني الأضلاع والأجسام الفقرية.
- أنت في خطر متزايد للإصابة بالكسور.
- لديك ألم في العظام.
- تصاب بهشاشة العظام.
الحديد الزائد
غالبًا ما يخزن الأشخاص المصابون بأشكال حادة من الثلاسيميا الكثير من الحديد في الجسم (الحديد الزائد) ، والذي لم يعد يتحلل من تلقاء نفسه. هذا يمكن أن يتلف الأعضاء (داء ترسب الأصبغة الدموية الثانوي). تأتي زيادة الحديد ، من بين أمور أخرى ، من الدم الزائد الذي يتلقاه الشخص المصاب بانتظام عن طريق الحقن.
بالإضافة إلى ذلك ، يعتبر الجسم فقر الدم على أنه نقص في الحديد ويحاول تعويض ذلك عن طريق تناول المزيد من الحديد من الطعام. ينتج الجسم أيضًا خلايا دم زائدة. ثم يعمل الكبد والطحال بأقصى سرعة لتفتيتهما مرة أخرى.
يتسبب تركيز الحديد المرتفع بشكل مفرط في ظهور أعراض أخرى لدى المصابين ، مثل:
- فشل القلب (قصور القلب)
- عدم انتظام ضربات القلب
- ضعف الكبد
- مرض السكري (داء السكري)
- قصر القامة
- تأخر البلوغ
- خمول الغدة الدرقية (قصور الغدة الدرقية)
- اضطرابات التمثيل الغذائي لفيتامين د
متى يجب زيارة الطبيب
نظرًا لأن الأنواع الشديدة من الثلاسيميا تظهر غالبًا في الأشهر القليلة الأولى من الحياة ، فمن المهم أن يرى الوالدان طبيب أطفال في أسرع وقت ممكن عند ظهور العلامات الأولى (مثل الشحوب).
كيف تستطيع منع ذالك؟
ينصح الأطباء الأشخاص الذين يعرفون أن لديهم الجين الخاص بالثلاسيميا بالذهاب إلى مركز الاستشارات الوراثية قبل التخطيط للحمل أو إذا كانوا يريدون إنجاب الأطفال.
يستخدم المتخصصون المدربون فحص الدم الجيني لتحديد المخاطر الجينية المحتملة المرتبطة بالحمل عند الأزواج.
يُنصح أيضًا الأشخاص الذين لديهم تاريخ عائلي معروف للإصابة بالثلاسيميا بطلب المشورة قبل الحمل.
لمنع الالتهابات التي تهدد الحياة ، يوصي الأطباء أيضًا بتطعيم الأطفال المصابين بالثلاسيميا والأطفال الأصحاء وفقًا لتقويم التطعيم الحالي. في بعض الحالات ، من الضروري علاج الأطفال المصابين بالثلاسيميا بانتظام بمضادات حيوية معينة. هذه تساعد في منع الالتهابات البكتيرية الخطيرة ، والتي يكون الأشخاص المصابون بالثلاسيميا أكثر عرضة لها بشكل خاص.
يساعد القياس المنتظم لدرجة حرارة الجسم على اكتشاف العدوى في أقرب وقت ممكن وعلاجها في أسرع وقت ممكن. إذا كانت لديك حمى تزيد عن 38.5 درجة مئوية ، يجب أن ترى الطبيب على الفور. من الممكن أن تكون العدوى هي سبب ذلك.
من المهم أيضًا بشكل خاص أن يقوم الطبيب بفحص المرضى بأنفسهم بانتظام. هذه هي الطريقة الوحيدة لمراقبة مسار المرض بعناية وتحديد المضاعفات وعلاجها في مرحلة مبكرة.
هل الثلاسيميا قابل للشفاء؟
لا يمكن حاليًا العلاج الكامل لمرض الثلاسيميا إلا من خلال زرع الخلايا الجذعية أو العلاج الجيني. ارتفع متوسط العمر المتوقع ونوعية المصابين بشكل مستمر بفضل الأدوية الجديدة وتحسين العلاج والتعليم الأفضل.
ما هو تشخيص مرض الثلاسيميا؟
الأشخاص المصابون بالثلاسيميا الخفيفة (ثلاسيميا ألفا أو بيتا الصغيرة) لديهم متوسط عمر متوقع طبيعي. كقاعدة عامة ، من الممكن أن يعيش المصابون دون إعاقة.
مع الأشكال الأكثر شدة ، غالبًا ما يكون من الممكن أيضًا تحقيق متوسط العمر المتوقع تقريبًا من خلال العلاج الصحيح والمبكر (مثل عمليات نقل الدم المنتظمة). ومع ذلك ، فإن العلاج المستمر والمكثف مدى الحياة ضروري.
في بعض الأحيان ، يمكن أن تعالج زراعة الخلايا الجذعية المرض ، وفي حالات نادرة ، العلاج الجيني.
كذا: أعراض طفل رضيع ولادة الحمل